


Curr. Oncol. 2024, 31(4), 2305-2315; https://doi.org/10.3390/curroncol31040171 (registering DOI) - 19 Apr 2024
Abstract
►
Show Figures
Background: pregnancy-associated breast cancer (PABC) affects one in 3000 pregnancies, often presenting with aggressive features. Methods: We retrospectively evaluated a cohort of 282 young BC patients (≤45 years old) treated between 1995 and 2019, dividing them into three groups: nulliparous women, women with
[...] Read more.
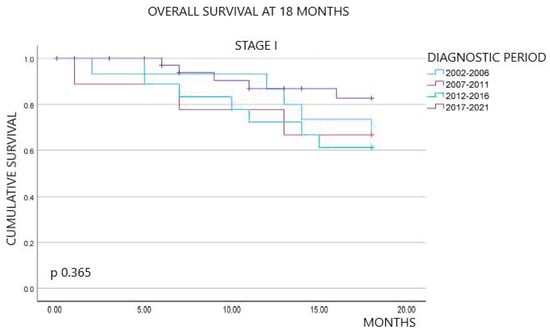
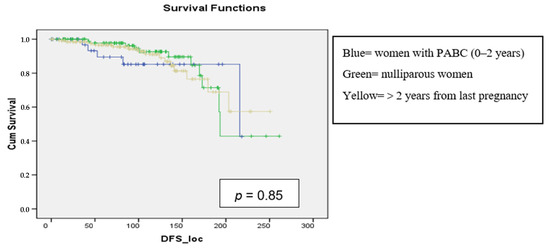
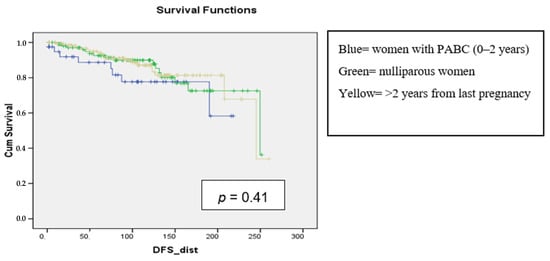
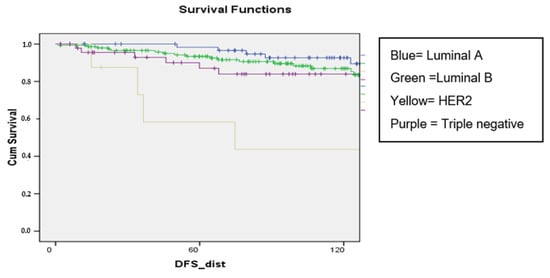
Background: pregnancy-associated breast cancer (PABC) affects one in 3000 pregnancies, often presenting with aggressive features. Methods: We retrospectively evaluated a cohort of 282 young BC patients (≤45 years old) treated between 1995 and 2019, dividing them into three groups: nulliparous women, women with PABC (diagnosed within 2 years since last pregnancy) and women with BC diagnosed > 2 years since last pregnancy. This last group was further stratified according to the time between pregnancy and BC. The analysis encompassed histological factors (tumor size, histotype, grading, nodal involvement, multifocality, lympho-vascular invasion, hormone receptor expression, Ki-67 index, and HER2 expression), type of surgery and recurrence. Results: Age at diagnosis was younger in nulliparous than in parous women (p < 0.001). No significant differences were noticed regarding histological characteristics and recurrences. At univariate analysis, nodal involvement (OR = 2.4; p < 0.0001), high tumor grade (OR = 2.6; p = 0.01), and lympho-vascular invasion (OR = 2.3; p < 0.05), but not pregnancy (OR = 0.8; p = 0.30), influenced DFS negatively. Multivariate analysis confirmed nodal involvement as the only negative independent prognostic factor for a worse DFS (OR = 2.4; p = 0.0001). Conclusions: in our experience, pregnancy is not an independent adverse prognostic factor for BC DFS.
Full article
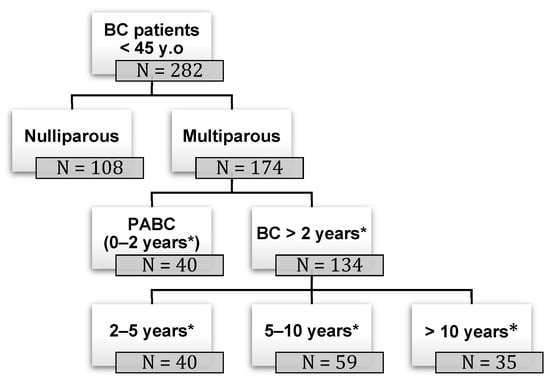
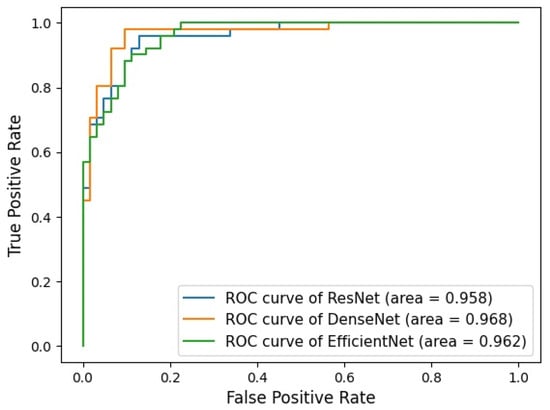
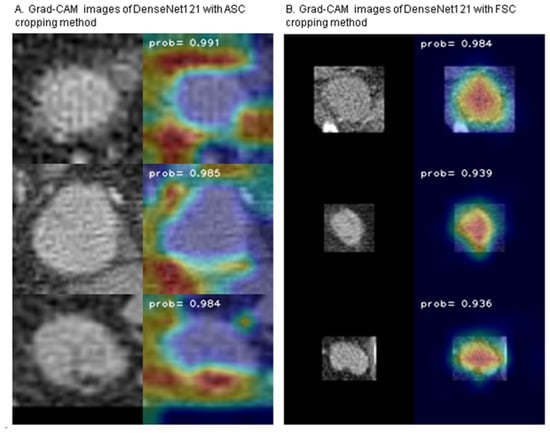
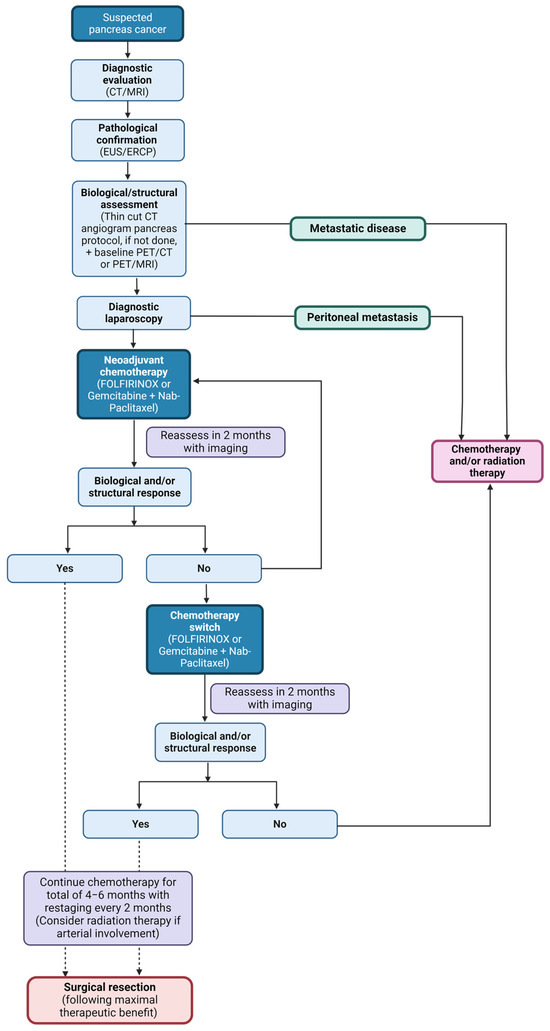
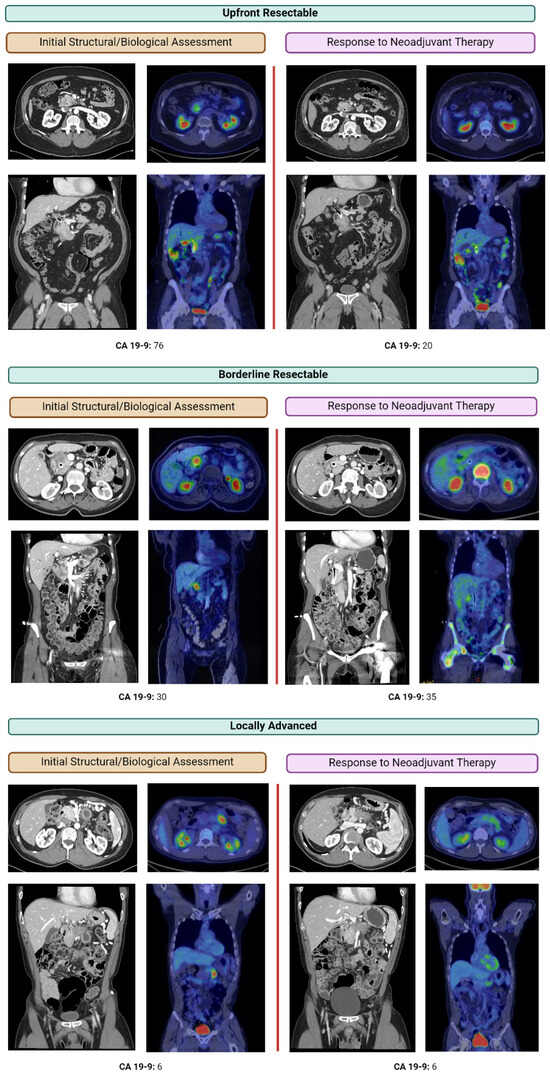
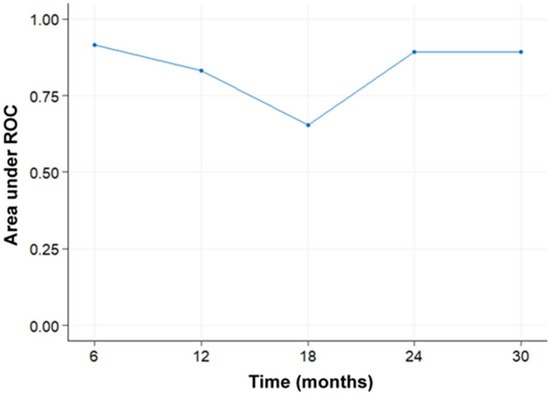
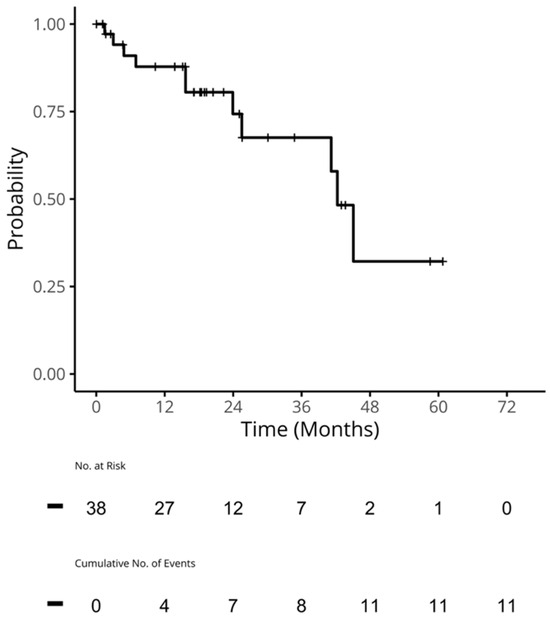
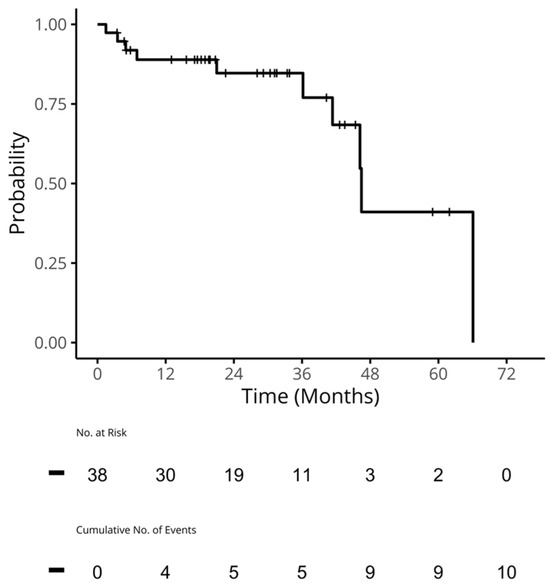
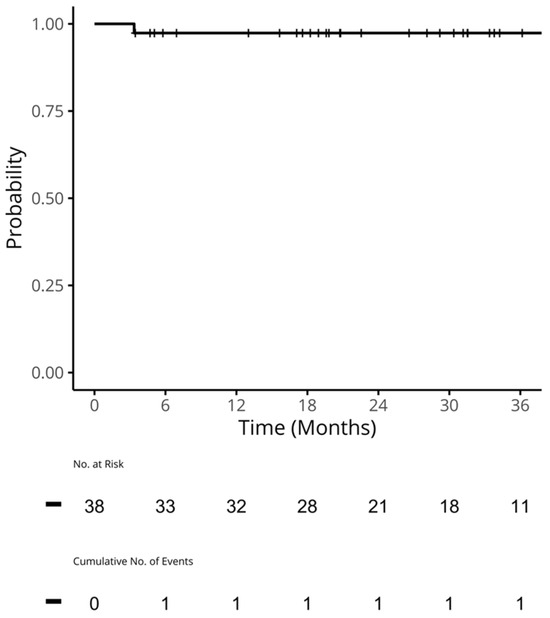
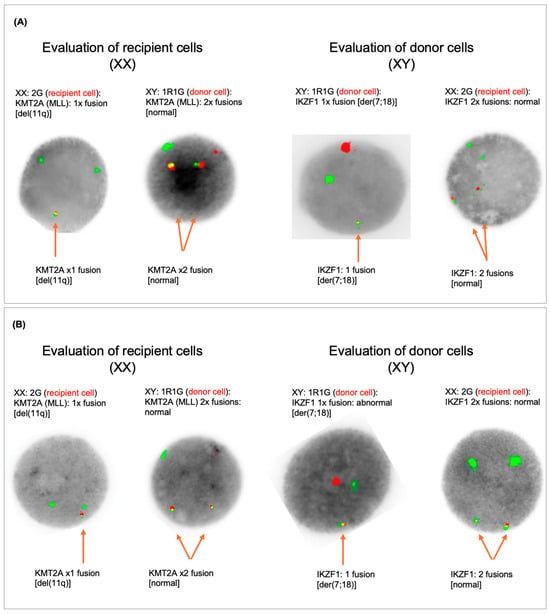
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}