


Curr. Oncol. 2024, 31(4), 2278-2288; https://doi.org/10.3390/curroncol31040169 - 18 Apr 2024
Abstract
►
Show Figures
Background: Accurate detection of axillary lymph node (ALN) metastases in breast cancer is crucial for clinical staging and treatment planning. This study aims to develop a deep learning model using clinical implication-applied preprocessed computed tomography (CT) images to enhance the prediction of ALN
[...] Read more.
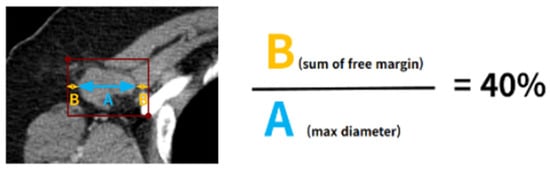
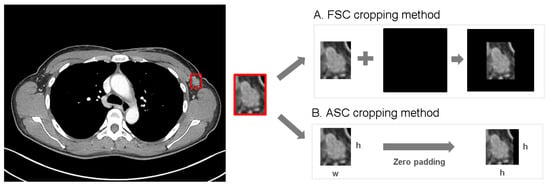
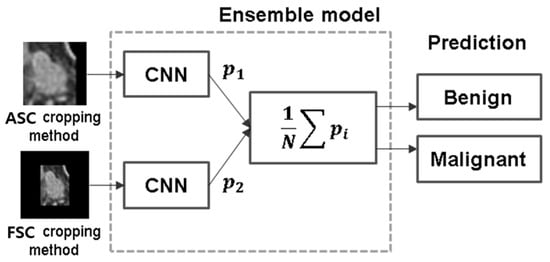
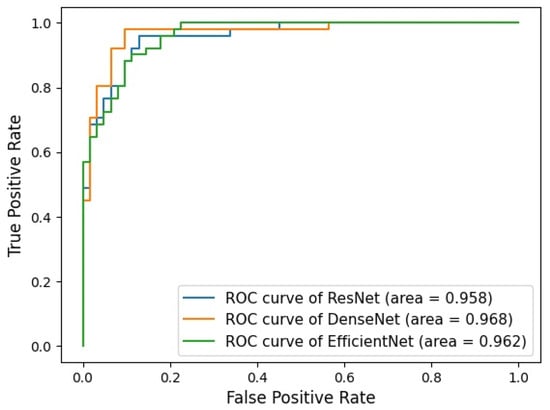
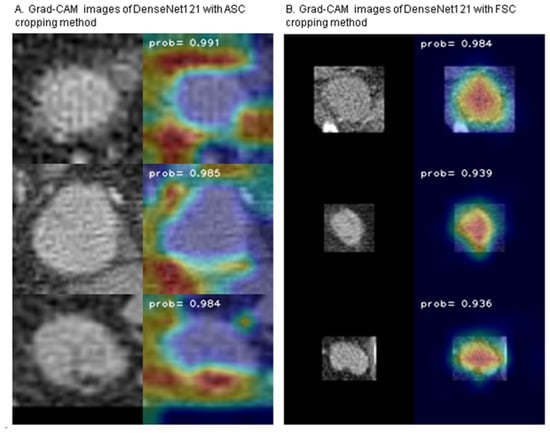
Background: Accurate detection of axillary lymph node (ALN) metastases in breast cancer is crucial for clinical staging and treatment planning. This study aims to develop a deep learning model using clinical implication-applied preprocessed computed tomography (CT) images to enhance the prediction of ALN metastasis in breast cancer patients. Methods: A total of 1128 axial CT images of ALN (538 malignant and 590 benign lymph nodes) were collected from 523 breast cancer patients who underwent preoperative CT scans between January 2012 and July 2022 at Hallym University Medical Center. To develop an optimal deep learning model for distinguishing metastatic ALN from benign ALN, a CT image preprocessing protocol with clinical implications and two different cropping methods (fixed size crop [FSC] method and adjustable square crop [ASC] method) were employed. The images were analyzed using three different convolutional neural network (CNN) architectures (ResNet, DenseNet, and EfficientNet). Ensemble methods involving and combining the selection of the two best-performing CNN architectures from each cropping method were applied to generate the final result. Results: For the two different cropping methods, DenseNet consistently outperformed ResNet and EfficientNet. The area under the receiver operating characteristic curve (AUROC) for DenseNet, using the FSC and ASC methods, was 0.934 and 0.939, respectively. The ensemble model, which combines the performance of the DenseNet121 architecture for both cropping methods, delivered outstanding results with an AUROC of 0.968, an accuracy of 0.938, a sensitivity of 0.980, and a specificity of 0.903. Furthermore, distinct trends observed in gradient-weighted class activation mapping images with the two cropping methods suggest that our deep learning model not only evaluates the lymph node itself, but also distinguishes subtler changes in lymph node margin and adjacent soft tissue, which often elude human interpretation. Conclusions: This research demonstrates the promising performance of a deep learning model in accurately detecting malignant ALNs in breast cancer patients using CT images. The integration of clinical considerations into image processing and the utilization of ensemble methods further improved diagnostic precision.
Full article
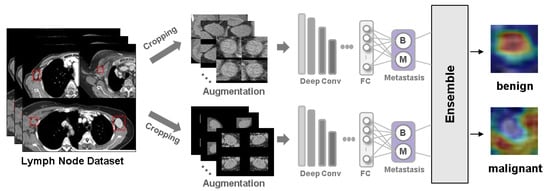
Figure 1








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}