


Curr. Oncol. 2024, 31(5), 2353-2363; https://doi.org/10.3390/curroncol31050175 (registering DOI) - 23 Apr 2024
Abstract
Myelodysplastic neoplasm (MDS) is a heterogeneous group of clonal hematological disorders that originate from the hematopoietic and progenitor cells and present with cytopenias and morphologic dysplasia with a propensity to progress to bone marrow failure or acute myeloid leukemia (AML). Genetic evolution plays
[...] Read more.
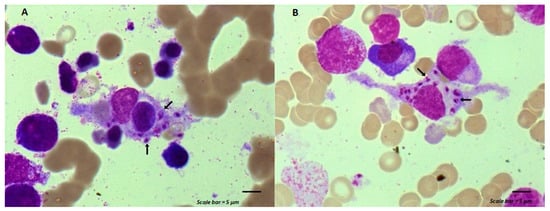
Myelodysplastic neoplasm (MDS) is a heterogeneous group of clonal hematological disorders that originate from the hematopoietic and progenitor cells and present with cytopenias and morphologic dysplasia with a propensity to progress to bone marrow failure or acute myeloid leukemia (AML). Genetic evolution plays a critical role in the pathogenesis, progression, and clinical outcomes of MDS. This process involves the acquisition of genetic mutations in stem cells that confer a selective growth advantage, leading to clonal expansion and the eventual development of MDS. With the advent of next-generation sequencing (NGS) assays, an increasing number of molecular aberrations have been discovered in recent years. The knowledge of molecular events in MDS has led to an improved understanding of the disease process, including the evolution of the disease and prognosis, and has paved the way for targeted therapy. The 2022 World Health Organization (WHO) Classification and the International Consensus Classification (ICC) have incorporated the molecular signature into the classification system for MDS. In addition, specific germline mutations are associated with MDS development, especially in pediatrics and young adults. This article reviews the genetic abnormalities of MDS in adults with a brief review of germline predisposition syndromes.
Full article
(This article belongs to the Special Issue The Molecular Pathology of Myelodysplastic Syndromes)
►
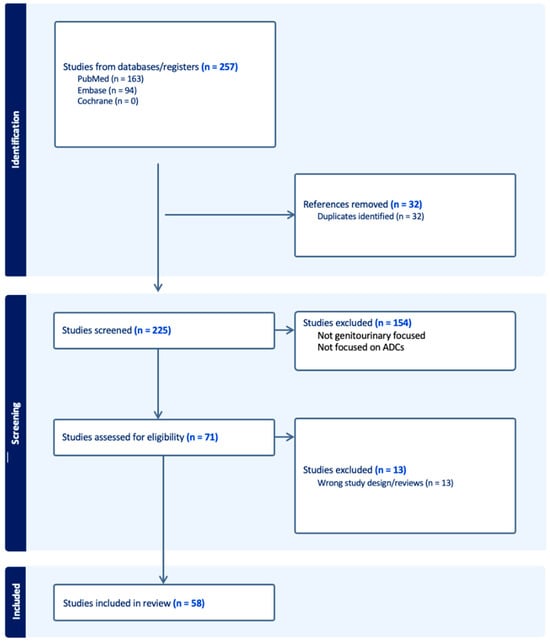
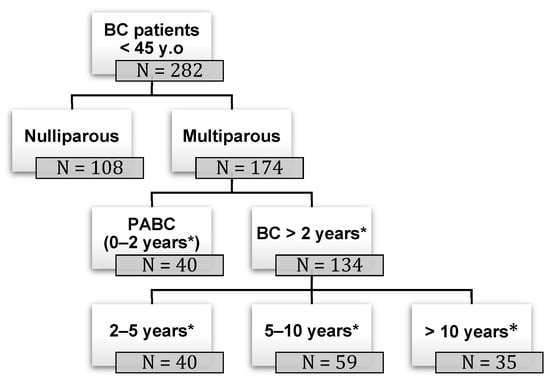
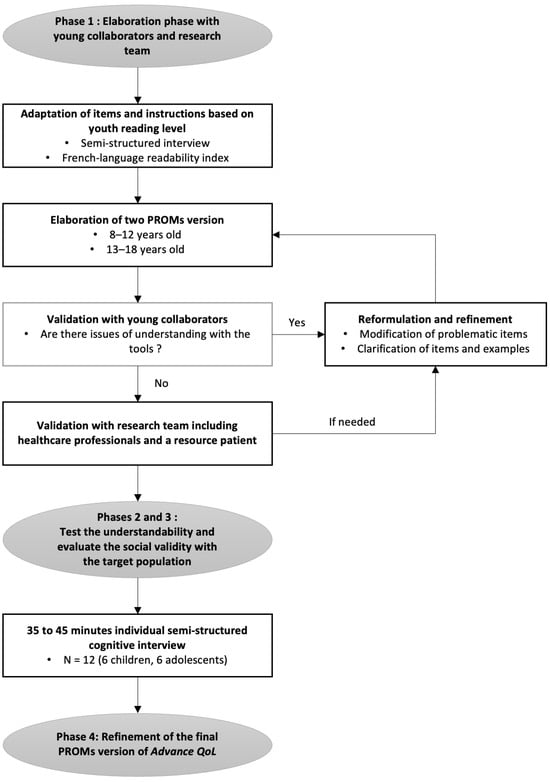
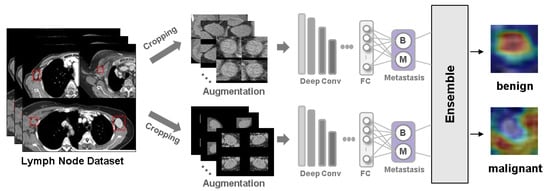
Show Figures
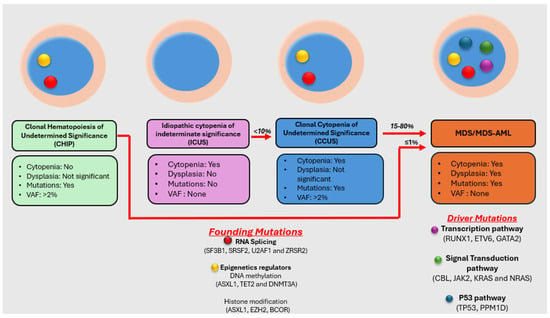
Figure 1





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}